Remembering Josephine:
Josephine Rose Lampitt

The Lampitt’s family story was featured in the February 2010 edition of The Health Journal. Read the article at The Health Journal online or download the PDF. Josephine’s story was also featured in the March 21, 2010 issue of the Daily Press.
Our story began in early December 2008. We were scheduled to bring our then eight-month-old daughter, Josephine Rose Lampitt, to Children’s Hospital of The King’s Daughters (CHKD) in Norfolk. She had been seen by other doctors before for what various specialists had repeatedly described as “severe reflux,” a relatively common and benign disorder that most children outgrow.
We expected our visit to include a continuation of the reflux diagnosis but also a more detailed read-out on the cause for her newer symptoms, among them poor weight gain, unusual eye movements and persistent crankiness. The weight gain issue in particular was perplexing as we had tried for months to increase Josephine’s body weight with breast milk supplements, then formula, and eventually a high-calorie formula concentrate. Nevertheless, we’d been assured that there was no reason to worry.
 To our horror, we soon found ourselves confronting every parent’s worst nightmare: a diagnosis of an extremely rare and incurable disease that would take our infant daughter from us. The doctors explained that Josephine had a rare metabolic disease— specifically, a lysosomal storage disorder known as Gaucher (pronounced “go-shay”) disease. Of the three forms of the disease, only Type I has a sustainable treatment while Types II and III have a degenerative neurological component that is untreatable and incurable. Josephine had the rarest form, Type II, which affects approximately one in 100,000 live births. The average life span, we were told, was about nine months, though some children had lived as long as two to three years.
To our horror, we soon found ourselves confronting every parent’s worst nightmare: a diagnosis of an extremely rare and incurable disease that would take our infant daughter from us. The doctors explained that Josephine had a rare metabolic disease— specifically, a lysosomal storage disorder known as Gaucher (pronounced “go-shay”) disease. Of the three forms of the disease, only Type I has a sustainable treatment while Types II and III have a degenerative neurological component that is untreatable and incurable. Josephine had the rarest form, Type II, which affects approximately one in 100,000 live births. The average life span, we were told, was about nine months, though some children had lived as long as two to three years.
Words fall short of capturing the emotions we felt then and have felt ever since, but shock, disbelief and horror are starting points. We had no known family history of that disease nor any disease for that matter. My wife and I had above-average health. We were both college-educated and from seemingly average American families. We were financially stable with a sound marriage and strong faith. We already had one healthy son. Until that time we naively believed that this was the type of tragedy that affected others, the proverbial “people down the street.” We could not have been more wrong.
 We learned that Gaucher disease passes down via mutated recessive genes (present in approximately one in 400 people in the general population). Neither my wife nor I had ever displayed symptoms, nor had any of our relatives. For symptoms of Gaucher disease to manifest, both parents must have the recessive gene, and even then, the couple’s offspring have only a one in four chance of manifesting the disease (each child also has a 50-percent chance of carrying the disease with no symptoms and a 25-percent chance of having no trace of the disorder).
We learned that Gaucher disease passes down via mutated recessive genes (present in approximately one in 400 people in the general population). Neither my wife nor I had ever displayed symptoms, nor had any of our relatives. For symptoms of Gaucher disease to manifest, both parents must have the recessive gene, and even then, the couple’s offspring have only a one in four chance of manifesting the disease (each child also has a 50-percent chance of carrying the disease with no symptoms and a 25-percent chance of having no trace of the disorder).
We soon found ourselves looking back fondly on Josephine’s first six months when she behaved and appeared, for the most part, as a normal, healthy baby girl. She had been irritable at points, but that in itself had not alarmed us. Likewise, she had raspy breathing, but that had been attributed to the reflux. She made unusual facial expressions from time to time, but we (and her doctors) had reminded ourselves that every baby has his or her own ‘isms,’ quirks and silly expressions. She had made our family complete—a mom, a dad, a son and then sweet Josephine to balance the gender mix. We had all been thrilled when she was born, especially our son, who was eager to see his little sister grow up before his eyes.
Now, my wife and I were faced with how to take care of a dying child, how to ensure Josephine had the best short little life possible with the best access to doctors, the most quality time with family possible and the most comfort science and faith could offer.
 We also had to consider our son’s emotional and mental health through her illness and, we feared, her impending death. My wife and I put our own needs—physical, mental, emotional—largely on hold, though we remained attuned to the fact that preserving our marriage during this time of chaos would be key to giving Josephine and our son the best support.
We also had to consider our son’s emotional and mental health through her illness and, we feared, her impending death. My wife and I put our own needs—physical, mental, emotional—largely on hold, though we remained attuned to the fact that preserving our marriage during this time of chaos would be key to giving Josephine and our son the best support.
Josephine’s doctors reminded us of the disease’s low incidence rate and admitted their lack of experience and comfort in treating children diagnosed with it. Dr. Virginia Proud, our pediatric genetic specialist at CHKD, admitted she had only seen one other case of Gaucher Type II in her career; our local pediatrician, Dr. Jennifer Altman, had seen none. We scoured the Web for every morsel of information we could find on Gaucher disease, Type II. We found very little.
We learned of a single organization dedicated specifically to Gaucher Type II and III—the Children’s Gaucher Disease Research Fund—through which we gained some insight but also fear and increased sadness. The group’s Website (www.childrensgaucher.org) conveyed the stories of children who had already succumbed to the disease. Each child’s story drilled home to us the reality of Josephine’s future.
 By mid-December 2008, Josephine started receiving all of her meals through a nasal-gastric tube inserted through her nose. This was the only means of increasing her caloric intake since the disease had begun to impair her ability to swallow and/or swallow without liquid entering her lungs. The lasting image in my mind of a tube protruding from our child’s nose is indescribable. Moreover, the mechanical and logistical challenges of administering meals and medications through the tube only added to our physical fatigue.
By mid-December 2008, Josephine started receiving all of her meals through a nasal-gastric tube inserted through her nose. This was the only means of increasing her caloric intake since the disease had begun to impair her ability to swallow and/or swallow without liquid entering her lungs. The lasting image in my mind of a tube protruding from our child’s nose is indescribable. Moreover, the mechanical and logistical challenges of administering meals and medications through the tube only added to our physical fatigue.
On Dec. 23, Josephine had her first round of enzyme replacement therapy (ERT). The treatments, which she would need two to three times a month, helped to ease some of her non-neurological symptoms such as an enlarged spleen and liver. We undertook the lengthy, costly therapies to improve the quality of her life as no treatment can stop or even stall the neurological degeneration caused by Gaucher. In the end, the ERT treatments cost tens of thousands of dollars, but we never once hesitated to incur the cost for ourselves or our insurance provider. (Our provider, Anthem, did not balk at a single expense.)
By January 2009 we decided to bring Josephine to one of the world’s premier institutions for medical research—the U.S. National Institutes of Health (NIH) outside of Washington, D.C. We were reminded in advance that NIH’s mission is to research treatments, but that the dividends of the research process would be our (and future generations’) better understanding of the disease. Clearly, our goals for Josephine had changed. We continued to hope for a miracle, but we conceded that modern science could not save our child. We sought information from researchers to help us enhance Josephine’s quality of life and, ultimately, quality of death. Though our NIH doctors proved to be a tremendous resource as they interpreted a battery of tests, they couldn’t predict how Josephine’s specific gene mutations would manifest themselves in the coming weeks or months.
 We decided we’d keep Josephine at home as much as possible, out of arm’s reach of doctors who openly admitted they could not help us. This decision, in effect, removed the uncertainty over an extremely long-shot, unproven option of a bone marrow transplant in North Carolina.
We decided we’d keep Josephine at home as much as possible, out of arm’s reach of doctors who openly admitted they could not help us. This decision, in effect, removed the uncertainty over an extremely long-shot, unproven option of a bone marrow transplant in North Carolina.
An oxygen pump and pulse-oxygen monitoring machine soon arrived at our home. The monitoring device measured the oxygen level in Josephine’s blood at night while she slept (and eventually during the day as her disease progressed). With increasing frequency, the device would emit a piercing alarm—indicating that her ever-weakening lungs couldn’t meet her body’s demands for oxygen. The alarms became so frequent that (with approval from doctors) we gradually adjusted the device’s sensitivity to allow us to sleep through some of her less-serious spells. Other nights were so rough that we took shifts sleeping on her floor huddled in a blanket. Being closer to Josephine allowed us to respond more quickly with a direct, focused administration of life-saving oxygen and, equally important, a warm, reassuring hug for our sick, panicked child.
In short, we went to bed each night not knowing whether Josephine would still be with us in the morning. It was terrifying.
 Through all our hospital visits, and especially the home visits of our warm-hearted and gifted pediatric nurse, Mr. Jaime Budy, we became increasingly well-versed in terms and concepts we had never before grappled with. We learned what a palliative care expert was, and we learned the names of medicines such as Ativan (for anxiety), Robinul (an expectorant) and morphine (for pain) and the proper method for administering these potent drugs around the clock. We came to understand the nuances of a Do Not Resuscitate (DNR) order, a legal document designed to clarify to emergency responders whether to give life-saving resuscitation.
Through all our hospital visits, and especially the home visits of our warm-hearted and gifted pediatric nurse, Mr. Jaime Budy, we became increasingly well-versed in terms and concepts we had never before grappled with. We learned what a palliative care expert was, and we learned the names of medicines such as Ativan (for anxiety), Robinul (an expectorant) and morphine (for pain) and the proper method for administering these potent drugs around the clock. We came to understand the nuances of a Do Not Resuscitate (DNR) order, a legal document designed to clarify to emergency responders whether to give life-saving resuscitation.
On a more mundane level, we had to walk past the children’s spring clothing lines at stores, knowing Josephine would likely not need new outfits. We had to stop saying “It will be OK” in response to our daughter’s cries when we knew in our hearts that it would not. More disturbing, but necessary, we had to consider how, when and where Josephine might die.
In the latter half of January, Josephine’s nasal-gastric tube was replaced with a tube that went directly to her stomach, giving her tender nose reprieve from the constant rubbing of the feeding tube and allowing her hands to swing more freely. Now that the feeding tube was hidden securely under her clothes, she could finally feel her face again without risk of pulling the tube out.
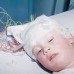
Due to increasingly frequent and alarming sleep disturbances, we admitted Josephine to CHKD in early February 2009 so that doctors could conduct an overnight sleep study and hopefully determine the cause. The study revealed that the disease was causing severe sleep apnea (obstructive and central) and depriving her of “active sleep,” which is similar to REM in adults. [Ed.: Obstructive sleep apnea is the result of a collapsed or blocked airway while the central type is neurological, meaning the brain stops telling the body to breathe.] It was during that visit that CHKD Sleep Expert Dr. Michael Dubik noted the precious manner in which electrode-peppered Josephine caressed her mother’s face. His passing comment inspired me to snap a photo (above). That photo tells our story; it motivates me to this day.
 We had a serious scare on Feb. 15. Whereas previous apneic episodes had occurred at night, Josephine experienced one in the middle of the day during naptime. By chance—the monitor was not set up for daytime alerts—we discovered her mid-nap, a pronounced shade of blue. We quickly administered the oxygen pump and seconds later were relieved to have her back in our arms as a smiling child. Nonetheless, we were reminded that she was living on borrowed time. We called nearby relatives for moral support and were comforted by their visit. We sent our son home with his cousins for a sleepover out of fear that Josephine would not make it through the night.
We had a serious scare on Feb. 15. Whereas previous apneic episodes had occurred at night, Josephine experienced one in the middle of the day during naptime. By chance—the monitor was not set up for daytime alerts—we discovered her mid-nap, a pronounced shade of blue. We quickly administered the oxygen pump and seconds later were relieved to have her back in our arms as a smiling child. Nonetheless, we were reminded that she was living on borrowed time. We called nearby relatives for moral support and were comforted by their visit. We sent our son home with his cousins for a sleepover out of fear that Josephine would not make it through the night.
To our surprise, the night was uneventful. Josephine woke up the next morning looking and feeling great. We cut up an apple and let her taste it. This was consistent with doctors’ advice to allow Josephine to enjoy one of life’s most basic pleasures. As with lollipops a few days before, and decaffeinated coffee and tea before that, she lit up with excitement. Such tastes would probably excite any child, but they took on special meaning for Josephine. By this point, she was not tasting any food or liquids; her nutrients were delivered straight to her stomach via a gastrointestinal tube.
After Shutterbug Dad snapped a few photos of Josephine licking the apple, we packed her up in the car and drove to pick up her brother at our relatives’ house. Since she was doing so well and we planned to return home shortly, we left her medicines and equipment at home. We had a good visit, and Josephine even had some pleasingly strong laughs and giggles as she tasted some tea from her mother’s mug.
 Monday, Feb. 16—President’s Day, I remember—was warm enough that the older children went outside to play. My wife laid Josephine down to change her diaper, and that’s when we noticed Josephine was getting short on breath. And then it happened. Despite weeks of nervous anticipation and inconsistent medical advice about how long she might live, Josephine was gone.
Monday, Feb. 16—President’s Day, I remember—was warm enough that the older children went outside to play. My wife laid Josephine down to change her diaper, and that’s when we noticed Josephine was getting short on breath. And then it happened. Despite weeks of nervous anticipation and inconsistent medical advice about how long she might live, Josephine was gone.
We had known this moment would come, and had made various preparations, but nothing could prepare us for the shock. We were allowed privacy as we said goodbye to Josephine, holding her in our arms for an hour or so before we made the dreaded phone calls. She escaped this world in a natural state, free from tubes and machines and hospitals and doctors. She cutely circumvented every man-made device designed to hold on to her, undermined every plan we’d made for her last days and minutes. She reminded us of those things we cannot control.
Josephine’s death marked an important crossroads in our struggle. Her weight shifted from our arms and into our hearts, where it has remained. But we view her death as a turning point, not an end. This is not to say we are not still grieving. Under any other circumstances we’d have an almost-two-year-old running around the house. We miss her giggles, her smiles and all her silly quirks. Thoughts of her fill our lives yet we feel her absence.
 In the months since Josephine’s death, bold acquaintances have asked if we feel her presence. Are there angels, and does she send us signs? Are there miracles? What else could explain the strong scent of flowers in the family van on the morning of her death and, in the months that followed, the mysterious workings of not one but two car radios, the inexplicable healing of my back pain after years of suffering, and too many rainbows over the house to count?
In the months since Josephine’s death, bold acquaintances have asked if we feel her presence. Are there angels, and does she send us signs? Are there miracles? What else could explain the strong scent of flowers in the family van on the morning of her death and, in the months that followed, the mysterious workings of not one but two car radios, the inexplicable healing of my back pain after years of suffering, and too many rainbows over the house to count?
It had always been our dream to have a larger family of three to four children. Doctors presented various means for us to have biological children with no risk of Gaucher disease, but those options were inconsistent with our beliefs. Likewise, we knew we would not consider terminating a pregnancy if our child tested positive. After countless hours of weighing the considerations, we decided to let go and open our hearts to the possibility of conceiving again. Our conclusion rested somewhat on the realization that, had we known of the defects in our genes years ago, we might have scared ourselves out of having our healthy son and even Josephine—whose impact on the world and people who knew her will outlive even us. Some may wonder whether Josephine’s short life was somehow not worth the pain, that perhaps she, too, would have preferred to not have experienced it all. We remind everyone that she experienced laughter and love from the first day until her very last. How many of us can really expect to be that fortunate?
Last July we learned that we would be parents once again—our son is due in early March. Many weeks were filled with uncertainty—but surprisingly, not fear—as we awaited the results of amniocentesis tests that would reveal whether our child in utero would display Gaucher disease. Last October we learned the results: negative. Like our older son, this child will not manifest symptoms.
As we approach the one-year anniversary of Josephine’s death (or, her “Angel Day” as we like to call it), we are confronted with an overlap of conflicting emotions. We are joyful and thankful for the expected arrival of our next son, but his arrival cannot “replace” Josephine, nor will we “move on” or “let go.” She’s still a part of our family. We talk about her, and to her, on a regular basis. We visit her grave as well as her still-intact bedroom to feel close to her. Photos of her are prominently displayed in our home. And when videos of Josephine play on a digital picture frame, our son runs towards it just to catch a glimpse of her giggling again.
At the same time, my wife must make snap decisions as to how much to tell strangers when they innocently ask about our baby-to-be, “Is this your second child?”, or comment, “Wouldn’t it be sweet to have a little girl?” While these remarks pierce our hearts, our hard-earned Ph.D. in life and death has made us largely unfazed by what others may consider important issues or major life stresses. We’ve lived through the worst hand life can deal, and we feel we can tackle anything new that comes our way. A year ago we felt utterly helpless; we feel invincible now. And that feeling of weathered strength allows us to leverage our suffering for a greater cause.
 To quote Greg Macres, founder of the Children’s Gaucher Disease Research Fund and father to the late Gregory Macres, lost 13 years ago to the disease:
To quote Greg Macres, founder of the Children’s Gaucher Disease Research Fund and father to the late Gregory Macres, lost 13 years ago to the disease:
“We can accept our pain and do nothing, or we can channel it to make the world a better place.”
Ed and Claire Lampitt
Williamsburg, Virginia
Site Search
Our 100% Commitment

Our Financial Commitment — 100% of every dollar we receive goes to medical research. How do we do this?
Combined Federal Campaign

"Children's Medical Charities of America" — CFC #76948. Federal Employees click here.
A Cure For Other Brain Diseases
Our research may help 20 other childhood diseases. There may also be an important connection between Gaucher Disease and Parkinson's.
CGRF Earns Seal of Excellence

CGRF has been honored for our high standards of accountability and effectiveness. Learn what the "Best in America" seal means.
Stay Connected

